
http://www.intramed.net/home.asp
La alteración de la secreción de insulina podría deberse a una disminución de la tasa de secreción celular o a una disminución de la masa de las células β o a ambos. Se analizan en profundidad los mecanismos íntimos del proceso.
Autor: James Cantley, Frances M. Ashcroft Fuente: BMC Biology (2015) 13:33 Insulin secretion and type 2 diabetes: why do β-cells fail?
¿Qué es la diabetes tipo 2?
La diabetes mellitus (DM) es un término que abarca una multitud de problemas con muchas etiologías, unificado por una característica común: la elevación patológica de la glucosa en la sangre. La hiperglucemia sostenida conduce daño tisular en los órganos susceptibles y finalmente provoca complicaciones, incluyendo la retinopatía, la nefropatía, la neuropatía periférica, la enfermedad cardiovascular y el accidente cerebrovascular.
Se calcula que hacia 2035 el número de afectados se duplicará. El notable aumento de la enfermedad en los últimos años no solo causa aflicción sino también una carga enorme y creciente en los sistemas de salud y la economía global. Muchos países gastan más del 10 % de su presupuesto sanitario en el tratamiento de la DM y sus complicaciones.
La DM tipo 2 (DM2) es la forma más común de la enfermedad, y es responsable de aproximadamente el 90% de los casos. Tiene un componente genético muy fuerte que es amplificado por factores como la edad, la obesidad, la dieta, la actividad física y el embarazo.
La DM2 se caracteriza por la secreción insuficiente de insulina por las células ß de los islotes pancreáticos, junto con la alteración de la acción de la insulina en el músculo, el hígado y la grasa (una condición denominada resistencia a la insulina). La hiperglucemia aparece cuando la secreción de insulina es incapaz de compensar la resistencia a la insulina. La resistencia a la insulina está aumentada en la obesidad, lo que explica, al menos en parte, porqué la obesidad aumenta el riesgo de DM2.
Por otra parte, hay formas hereditarias monogénicas raras que suelen presentarse a temprana edad, y son responsables del 1-2% de los casos de diabetes. A diferencia de la DM2, en la que habría múltiples genes que contribuyen a la enfermedad, la diabetes monogénica está causada por mutaciones en un solo gen. Muchos de esos genes codifican los reguladores de la transcripción, las enzimas metabólicas y los canales iónicos que regulan el acoplamiento estímulo-secreción de las células β, o pueden afectar el desarrollo del páncreas. Se destaca que las variantes genéticas comunes en muchos de los genes conocidos como causantes de la diabetes monogénica aumentan el riesgo de DM2; por lo tanto, su estudio puede ayudar a aclarar la etiología de la DM2.
La DM1 debe tratarse con inyecciones de insulina, debido a la falta de células β.
El tratamiento inicial de la DM2 consiste en modificaciones de la alimentación y del estilo de vida, seguidas de la administración de agentes hipoglucemiantes, lo cual puede aumentar la secreción de insulina (por ej., las sulfonilureas) o reducir la resistencia a la insulina o la producción de glucosa hepática (por ej., la metformina). Si estas medidas no logran controlar la hiperglucemia, entonces está indicada la insulina.
La diabetes monogénica debe tratarse de diferentes maneras, de acuerdo al gen involucrado.
¿Porqué no hay otras hormonas que puedan reemplazar a la insulina?
¿Hasta qué punto la diabetes tipo 2 es una enfermedad genética?
El riesgo de que una persona desarrolle DM2 está determinado por una compleja interacción entre la genética y el medio ambiente y los factores del estilo de vida. El genotipo desempeña claramente un papel genético: estudios prospectivos de gemelos monocigotas han mostrado una tasa de concordancia del 76% para la DM2 y de un 96% para la intolerancia a la glucosa.
La alteración de la secreción de insulina podría deberse a una disminución de la tasa de secreción celular o a una disminución de la masa de las células β o a ambos. Se analizan en profundidad los mecanismos íntimos del proceso.
Autor: James Cantley, Frances M. Ashcroft Fuente: BMC Biology (2015) 13:33 Insulin secretion and type 2 diabetes: why do β-cells fail?
¿Qué es la diabetes tipo 2?
La diabetes mellitus (DM) es un término que abarca una multitud de problemas con muchas etiologías, unificado por una característica común: la elevación patológica de la glucosa en la sangre. La hiperglucemia sostenida conduce daño tisular en los órganos susceptibles y finalmente provoca complicaciones, incluyendo la retinopatía, la nefropatía, la neuropatía periférica, la enfermedad cardiovascular y el accidente cerebrovascular.
Se calcula que hacia 2035 el número de afectados se duplicará. El notable aumento de la enfermedad en los últimos años no solo causa aflicción sino también una carga enorme y creciente en los sistemas de salud y la economía global. Muchos países gastan más del 10 % de su presupuesto sanitario en el tratamiento de la DM y sus complicaciones.
La DM tipo 2 (DM2) es la forma más común de la enfermedad, y es responsable de aproximadamente el 90% de los casos. Tiene un componente genético muy fuerte que es amplificado por factores como la edad, la obesidad, la dieta, la actividad física y el embarazo.
La DM2 se caracteriza por la secreción insuficiente de insulina por las células ß de los islotes pancreáticos, junto con la alteración de la acción de la insulina en el músculo, el hígado y la grasa (una condición denominada resistencia a la insulina). La hiperglucemia aparece cuando la secreción de insulina es incapaz de compensar la resistencia a la insulina. La resistencia a la insulina está aumentada en la obesidad, lo que explica, al menos en parte, porqué la obesidad aumenta el riesgo de DM2.
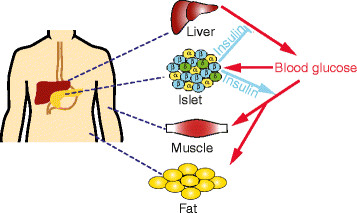
Homeostasis de la glucosa
- El aumento de la glucosa en la sangre provoca la secreción de
insulina de las células β que se hallan dentro de los islotes
pancreáticos.
- La insulina disminuye la glucosa en la sangre actuando sobre los
tejidos diana, suprimiendo la producción de glucosa por el hígado y
estimulando la captación de glucosa en el músculo y grasa.
- Las células α de los islotes pancreáticos son secretoras de glucagón.
- Las células δ de los islotes pancreáticos secretan somatostatina.
Por otra parte, hay formas hereditarias monogénicas raras que suelen presentarse a temprana edad, y son responsables del 1-2% de los casos de diabetes. A diferencia de la DM2, en la que habría múltiples genes que contribuyen a la enfermedad, la diabetes monogénica está causada por mutaciones en un solo gen. Muchos de esos genes codifican los reguladores de la transcripción, las enzimas metabólicas y los canales iónicos que regulan el acoplamiento estímulo-secreción de las células β, o pueden afectar el desarrollo del páncreas. Se destaca que las variantes genéticas comunes en muchos de los genes conocidos como causantes de la diabetes monogénica aumentan el riesgo de DM2; por lo tanto, su estudio puede ayudar a aclarar la etiología de la DM2.
La DM1 debe tratarse con inyecciones de insulina, debido a la falta de células β.
El tratamiento inicial de la DM2 consiste en modificaciones de la alimentación y del estilo de vida, seguidas de la administración de agentes hipoglucemiantes, lo cual puede aumentar la secreción de insulina (por ej., las sulfonilureas) o reducir la resistencia a la insulina o la producción de glucosa hepática (por ej., la metformina). Si estas medidas no logran controlar la hiperglucemia, entonces está indicada la insulina.
La diabetes monogénica debe tratarse de diferentes maneras, de acuerdo al gen involucrado.
¿Porqué no hay otras hormonas que puedan reemplazar a la insulina?
"Hay varias hormonas que pueden elevar la glucemia pero solo la insulina puede reducirla"
La mayoría de los sistemas de control, incluidos los fisiológicos, son superabundantes
(redundancia), lo que asegura que cuando un sistema falla otro se hace
cargo de la función. Por ejemplo, hay varias hormonas que pueden elevar
la glucemia pero solo la insulina puede reducirla. Al principio, esto
puede parecer sorprendente, pero vale la pena recordar que el exceso de
insulina tiene efectos mucho más inmediatos y deletéreos que poca
cantidad de insulina. Si la glucemia cae por debajo de 37 mg/dl en
tan solo 5 minutos, se puede producir daño cerebral letal. Por el
contrario, las complicaciones de la DM solo se producen cuando la
glucemia está crónicamente elevada durante muchas semanas y meses,
debido a una carencia sostenida de insulina. Por lo tanto, la hormona
fundamental es la insulina, la que es peligrosa tanto por exceso como
por defecto. Pero aunque se presta mucha atención a la falta de
insulina, y por consiguiente a la diabetes, el exceso agudo de insulina
es mucho más perjudicial.
"El exceso de insulina tiene efectos mucho más inmediatos y deletéreos que poca cantidad de insulina"
Otras funciones de la insulina─como su
capacidad para mejorar el crecimiento─se reflejan a través de varias
hormonas, como los factores de crecimiento símil insulina 1 y 2.
Solamente el papel de la insulina en la homeostasis de la glucosa es único. Por lo tanto, los autores especulan que la razón por la cual la capacidad de la insulina es única es el peligro de hipoglucemia, ya que para reducir la glucosa en sangre actúa a través de un receptor único.
En la historia evolutiva de los seres
humanos, cuando se enfrentaron a la comida inadecuada y al ejercicio no
planificado (escape de los depredadores) era más probable la hipoglucemia
que la hiperglucemia. En esta situación, dicen, es ventajoso tener una
única vía para descender la glucemia, ya que hay menos posibilidad de
hipoglucemia inadvertida.
Por el contrario, el hecho de que existan
numerosos sistemas de retroalimentación para reforzar el azúcar en
sangre es beneficioso. Aunque la DM2 es un problema creciente en las
sociedades actuales, en términos evolutivos es de poca importancia
porque generalmente se presenta después de la edad reproductiva
del individuo. Por otra parte, solo es muy reciente la exposición de
los seres humanos a la abundante disponibilidad de dietas hipercalóricas
y al sedentarismo, factores que impulsan la obesidad y la DM2.
¿Cómo hacen las células β para evitar la secreción inapropiada de insulina?
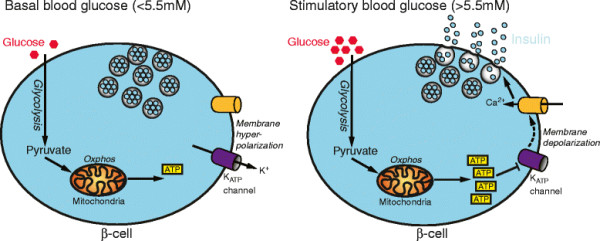
Las células β han evolucionado sus características metabólicas importantes para evitar la excesiva secreción de insulina y la hipoglucemia, especialmente durante el ejercicio. En primer lugar, la secreción de insulina es exquisitamente sensible a los cambios en la glucosa en sangre. Esto se logra porque el metabolismo de la glucosa se acopla a la secreción de insulina a través de los cambios en los niveles intracelulares de ATP, la actividad eléctrica de las células ß y la liberación de vesículas insulina.
¿Cómo hacen las células β para evitar la secreción inapropiada de insulina?
Las células β han evolucionado sus características metabólicas importantes para evitar la excesiva secreción de insulina y la hipoglucemia, especialmente durante el ejercicio. En primer lugar, la secreción de insulina es exquisitamente sensible a los cambios en la glucosa en sangre. Esto se logra porque el metabolismo de la glucosa se acopla a la secreción de insulina a través de los cambios en los niveles intracelulares de ATP, la actividad eléctrica de las células ß y la liberación de vesículas insulina.
En presencia de hiperglucemia,
la mayor parte de la glucosa absorbida por las células β se metaboliza a
través de la fosforilación oxidativa, elevando el ATP intracelular.
Esto cierra los canales KATP, desencadenando la actividad eléctrica de
las células β y provocando la afluencia de calcio (a través de los
canales de calcio voltaje dependientes) que, a su vez, estimulan la
liberación de insulina.
A la inversa, cuando los niveles de glucosa en sangre caen, la secreción de insulina se detiene rápidamente debido a la reducción del ATP intracelular en las células β, lo que lleva a la apertura de los canales KATP, la hiperpolarización de la membrana, la entrada de calcio reducido y, por lo tanto, la inhibición de la secreción de insulina.
En segundo lugar, una serie de genes metabólicos que están ampliamente expresados en otros tejidos no se expresan en las células β pancreáticas. Estos genes "no permitidos" son los que codifican la lactato deshidrogenasa y el transportador de β 1 (MCT1/SLC16A1), involucrados en el metabolismo del lactato y el piruvato. Esto evita la secreción de insulina en respuesta a la circulación de lactato y piruvato durante el ejercicio.
A la inversa, cuando los niveles de glucosa en sangre caen, la secreción de insulina se detiene rápidamente debido a la reducción del ATP intracelular en las células β, lo que lleva a la apertura de los canales KATP, la hiperpolarización de la membrana, la entrada de calcio reducido y, por lo tanto, la inhibición de la secreción de insulina.
En segundo lugar, una serie de genes metabólicos que están ampliamente expresados en otros tejidos no se expresan en las células β pancreáticas. Estos genes "no permitidos" son los que codifican la lactato deshidrogenasa y el transportador de β 1 (MCT1/SLC16A1), involucrados en el metabolismo del lactato y el piruvato. Esto evita la secreción de insulina en respuesta a la circulación de lactato y piruvato durante el ejercicio.
Las mutaciones en el gen SLC16A1, lo que motivan su expresión aberrante en las células β, provocan hipoglucemia inducida
por el ejercicio, permitiendo la secreción de insulina inducida por el
piruvato. En los primeros seres humanos, la hipoglucemia inducida por el
ejercicio podría ser letal, ya que impediría escapar de un depredador;
la ausencia de MCT1 asegura que la secreción de insulina permanezca
detenida durante el ejercicio. Del mismo modo, la adrenalina
inhibe la secreción de insulina, garantizando que los niveles de
glucemia no desciendan durante el ejercicio o en la respuesta de "lucha o
huida".
¿Qué ocasiona la deficiencia de insulina en la diabetes tipo 2?
La alteración de la secreción de insulina que se encuentra en la DM2 podría deberse a una disminución de la tasa de secreción celular (es decir, la función individual de las células β), o a una disminución de la masa de las células β (el producto del tamaño de las células β por su número), o ambos. Si bien ha habido mucho debate acerca de las contribuciones de la disfunción secretora y la pérdida de la masa de las células β relacionada con la secreción de insulina alterada en la DM2, todavía falta arribar a un consenso.
La alteración de la secreción de insulina que se encuentra en la DM2 podría deberse a una disminución de la tasa de secreción celular (es decir, la función individual de las células β), o a una disminución de la masa de las células β (el producto del tamaño de las células β por su número), o ambos. Si bien ha habido mucho debate acerca de las contribuciones de la disfunción secretora y la pérdida de la masa de las células β relacionada con la secreción de insulina alterada en la DM2, todavía falta arribar a un consenso.
Esto puede, en parte, deberse a la
dificultad para obtener islotes humanos de calidad y cantidad suficiente
para estudios funcionales (especialmente de donantes DM2), como los
programas de aislamiento de islotes destinados principalmente a
proporcionar islotes de donantes sanos para la terapia de trasplante.
Por otra parte, hay varios factores que
pueden variar entre las preparaciones de islotes humanos, confundiendo
así la comparación de la función de los islotes de DM2 con los
controles: Los donantes pueden haber recibido diferentes combinaciones
de fármacos antes de la muerte, y los factores genéticos y ambientales
pueden estar mal controlados. También las variaciones en el tiempo de
isquemia fría a la cual los islotes son expuestos durante el transporte
del páncreas y la aislación de los islotes, pueden alterar la expresión y
función de los genes.
Sin embargo, cuando muchas de las variables
anteriores son controladas, estudios con muestras de pequeño tamaño (n =
5 a 17 casos) han demostrado claramente que la secreción de insulina
estimulada por la glucosa (SIEG) es defectuosa en los
islotes de los donantes con DM2, comparado con los donantes no
diabéticos. En 2 de estos estudios, los islotes de los donantes DM2
respondieron normalmente a diferentes estímulos de la glucosa, lo que
sugiere una SIEG defectuosa en estas cohortes, probablemente debida más a
la sensibilización a la glucosa alterada (acoplamiento de la secreción-
estímulo) que a la pérdida del contenido de insulina o a un defecto
constitucional en la exocitosis de la insulina. Sin embargo, se
requieren más investigaciones para aumentar el número de casos
estudiados y estudiar en detalle la naturaleza de la respuesta SIEG
defectuosa.
Los estudios histológicos de la masa de células ß son más sencillos porque pueden ser realizados con tejidos fijados. Varios estudios han informado un descenso de la masa de células ß en la DM2. Sin embargo, una salvedad importante para estos experimentos es que las células ß suelen identificarse mediante la tinción de la insulina. Esto significa que el contenido de insulina debe ser suficientemente elevado como para detectarlas histológicamente─no se podrá hacer el recuento de células ß con un contenido de insulina muy reducido y por lo tanto, la masa de células ß será subestimada.
Los estudios histológicos de la masa de células ß son más sencillos porque pueden ser realizados con tejidos fijados. Varios estudios han informado un descenso de la masa de células ß en la DM2. Sin embargo, una salvedad importante para estos experimentos es que las células ß suelen identificarse mediante la tinción de la insulina. Esto significa que el contenido de insulina debe ser suficientemente elevado como para detectarlas histológicamente─no se podrá hacer el recuento de células ß con un contenido de insulina muy reducido y por lo tanto, la masa de células ß será subestimada.
Estudios recientes indican que los islotes
de diabéticos tipo 2 contienen muchas células ß que pueden ser
identificadas como tales utilizando la microscopia electrónica, por sus
característicos gránulos “huevo poché”, pero donde los gránulos son muy
escasos y la insulina es indetectable por inmunotinción. La
hiperglucemia produce efectos similares en un modelo de diabetes en
ratón. Por lo tanto, la medida que la masa de células ß está reducida en
la DM2 sigue siendo desconocida.
Si bien existe una buena evidencia de que la
tinción de la insulina de los islotes disminuye con el tiempo, todavía
se desconocen cuáles son las contribuciones relativas de la disminución
del contenido de insulina, el menor número de células β y el deterioro
del acoplamiento secreción-estímulo, en la menor secreción de insulina
en la DM2. A pesar de esto, en el período de 5 años posteriores al
diagnóstico, los pacientes con DM2 muestran una reducción del 25%
en la masa de células insulinpositivas, en relación con los controles
no diabéticos, mientras que en las en personas con DM2 de larga data
(>15 años), la masa de las células β se reduce en más del 50%. Esta
pérdida progresiva de la masa de células β durante la progresión de la
enfermedad supone una carga secretora cada vez mayor sobre las células β
que siguen siendo funcionales. Su resistencia probablemente esté
determinada por una interacción compleja entre el medio ambiente y los
factores genéticos y epigenéticos.
¿Los cambios en la identidad de las células β contribuyen a la diabetes tipo 2?
Es evidente que en el desarrollo de la DM2 hay múltiples mecanismos involucrados. Sin embargo, la evidencia reciente indica que la identidad de las células β no puede ser fija, y que los cambios en su identidad pueden contribuir a la secreción defectuosa de la insulina, dando lugar a la DM2.
¿Los cambios en la identidad de las células β contribuyen a la diabetes tipo 2?
Es evidente que en el desarrollo de la DM2 hay múltiples mecanismos involucrados. Sin embargo, la evidencia reciente indica que la identidad de las células β no puede ser fija, y que los cambios en su identidad pueden contribuir a la secreción defectuosa de la insulina, dando lugar a la DM2.
Está bien establecido que en los ratones, la
hiperglucemia da lugar a la expresión alterada de los factores de
transcripción de las células β y a la secreción defectuosa de la
insulina, una situación que se describe como desdiferenciación de las células β.
Estudios importantes recientes han
demostrado que en los ratones, la supresión de ciertos factores de
transcripción como FoxO1 lleva a la desdiferenciación de las células ß
del páncreas, que pierden su contenido de insulina y revierten a células
símil progenitoras de los islotes.
Del mismo modo, en un modelo de ratón
diabético se ha confirmado la expresión del marcador de células
progenitoras Ngn3. Pero si este es el caso, no está claro si lo mismo
sucede en las células β humanas en la DM2. Sin embargo, en la DM2 de los
seres humanos se han observado marcados cambios en los factores de
transcripción de las células β como así en los primates no humanos con
prediabetes inducida por la dieta.
Está bien establecido que la pérdida de la inmunotinción de insulina observada en muchos modelos de ratón diabético es paralela al aumento en la inmunotinción del glucagón. Estos cambios parecen estar estimulados por la hiperglucemia. En uno de estos modelos de ratón, el seguimiento del linaje reveló que un pequeño número de células β comienza a expresar glucagón. No obstante, es poco claro si esas células ß se convierten totalmente en células α, o si representan un tipo de célula intermedia que expresa glucagón como así muchas proteínas de las células ß (excepto la insulina).
Está bien establecido que la pérdida de la inmunotinción de insulina observada en muchos modelos de ratón diabético es paralela al aumento en la inmunotinción del glucagón. Estos cambios parecen estar estimulados por la hiperglucemia. En uno de estos modelos de ratón, el seguimiento del linaje reveló que un pequeño número de células β comienza a expresar glucagón. No obstante, es poco claro si esas células ß se convierten totalmente en células α, o si representan un tipo de célula intermedia que expresa glucagón como así muchas proteínas de las células ß (excepto la insulina).
Por el contrario, el seguimiento del linaje también mostró que tanto las células α como las células δ pueden convertirse en células β totalmente funcionantes, destacando la potencial plasticidad
de las células de los islotes. Por lo tanto, muchas investigaciones
actuales están dedicadas a conocer cómo se diferencian las células ß a
partir de las células progenitoras y otros tipos de células de los
islotes. Se destaca que los efectos de la hiperglucemia sobre
la diferenciación de las células ß, la pérdida del contenido de
insulina y la expresión de glucagón pueden revertirse con un estricto
control de la glucosa.
¿Lleva la obesidad a la insuficiencia de las células β?
Las epidemias actuales de obesidad y DM2 en todo el mundo muestran una tendencia y distribución geográfica notablemente similar. Hay evidencia importante de que el riesgo de DM2 aumenta con la obesidad. Sin embargo, la obesidad parece ejercer su efecto principalmente más en la resistencia a la insulina que en la función de las células β, y solo una minoría de las personas obesas desarrollará DM2, mientras que muchos individuos no obesos sí lo harán.
¿Lleva la obesidad a la insuficiencia de las células β?
Las epidemias actuales de obesidad y DM2 en todo el mundo muestran una tendencia y distribución geográfica notablemente similar. Hay evidencia importante de que el riesgo de DM2 aumenta con la obesidad. Sin embargo, la obesidad parece ejercer su efecto principalmente más en la resistencia a la insulina que en la función de las células β, y solo una minoría de las personas obesas desarrollará DM2, mientras que muchos individuos no obesos sí lo harán.
Por otra parte, la obesidad se asocia con una mejor respuesta insulínica a
la glucosa en individuos sin diabetes, mientras que estudios
histológicos recientes revelaron que la obesidad se asocia con un aumento
del 50% de la masa de células β. Así pues, parece que aparte de que la
obesidad causa insuficiencia de las células β, en algunas personas la
capacidad funcional de esas células para adaptarse a la obesidad falla,
lo que resulta en DM2.
| En los últimos 60 años hubo un dramático aumento de las tasas de DM2, que claramente no puede deberse a un cambio genético sino que se asocia con alteraciones en la dieta y el comportamiento, incluyendo un estilo de vida más sedentario y el aumento del consumo de alimentos hipercalóricos. |
¿Hasta qué punto la diabetes tipo 2 es una enfermedad genética?
El riesgo de que una persona desarrolle DM2 está determinado por una compleja interacción entre la genética y el medio ambiente y los factores del estilo de vida. El genotipo desempeña claramente un papel genético: estudios prospectivos de gemelos monocigotas han mostrado una tasa de concordancia del 76% para la DM2 y de un 96% para la intolerancia a la glucosa.
Por otra parte, los antecedentes familiares de DM 2 entrañan más del doble
de riesgo de que una persona desarrolle la enfermedad. Pero al mismo
tiempo, la evidencia epidemiológica muestra que en los últimos 60 años
hubo un dramático aumento de las tasas de DM2, que claramente no puede deberse
a un cambio genético sino que se asocia con alteraciones en la dieta y
el comportamiento, incluyendo un estilo de vida más sedentario y el
aumento del consumo de alimentos hipercalóricos.
El riesgo de DM2 también puede estar influenciado por cambios epigenéticos, que son alteraciones hereditarias que afectan la función de las células y que no implican cambios en la secuencia del ADN. Estos cambios epigenéticos están en gran parte determinados por factores ambientales, como la nutrición parental.
El riesgo de DM2 también puede estar influenciado por cambios epigenéticos, que son alteraciones hereditarias que afectan la función de las células y que no implican cambios en la secuencia del ADN. Estos cambios epigenéticos están en gran parte determinados por factores ambientales, como la nutrición parental.
La evidencia reciente sugiere que las células ß de los pacientes con DM2 tienen una alteración de la metilación
del ADN (una marca epigenética común) con cambios en los perfiles de la
expresión génica. Los estudios en roedores han demostrado que la
nutrición materna o paterna subóptima puede influir en las
modificaciones de la cromatina y la expresión génica en las células β de
la descendencia, consistente con la transmisión epigenética. Se sabe
que en los seres humanos, la nutrición materna y en los primeros años de
vida influye en el riesgo de DM2 de la descendencia. Se necesitan más
estudios para aclarar el papel emergente de la epigenética en la
etiología de la DM2.
¿Hay variantes de genes específicos asociadas a la diabetes tipo 2?
Esta pregunta no es fácil de responder. La DM2 es una enfermedad poligénica, y la evidencia actual apoya la idea de que en la mayoría de los individuos, el riesgo de desarrollar la enfermedad está determinado por la combinación del riesgo de variantes en muchos loci de genes, cada uno de los cuales confiere solo un pequeño aumento en el riesgo de enfermedad.
¿Hay variantes de genes específicos asociadas a la diabetes tipo 2?
Esta pregunta no es fácil de responder. La DM2 es una enfermedad poligénica, y la evidencia actual apoya la idea de que en la mayoría de los individuos, el riesgo de desarrollar la enfermedad está determinado por la combinación del riesgo de variantes en muchos loci de genes, cada uno de los cuales confiere solo un pequeño aumento en el riesgo de enfermedad.
Esto diferencia la DM2 de las formas de
diabetes monogénicas, mucho más raras, como la diabetes juvenil de
comienzo en la madurez (MODY) y la diabetes neonatal. También indica que
la DM2 es una entidad única, ya que la hiperglucemia
puede estar ocasionada por diferentes combinaciones de genes en
diferentes individuos, lo que también puede resultar en variaciones
fenotípicas.
El mejor método actual para identificar los genes que contribuyen a las enfermedades poligénicas son los estudios de asociación de todo el genoma, basados en la asociación de variantes genéticas comunes─polimorfismos de un solo nucleótido─con un fenotipo determinado, como la hiperglucemia. Hasta la fecha, en estudios de grandes cohortes se han encontrado más de 70 loci de genes que se asocian con la DM2, los que en su mayoría están implicados en la función de las células β.
Uno de los problemas en los estudios antes mencionados es el tamaño de las cohortes que deben ser estudiadas (a veces >100.000 personas) para generar suficiente poder estadístico. Tales cohortes son difíciles de fenotipificar con suficiente profundidad para revelar la compleja fisiología subyacente de la DM2. Así, inevitablemente, la mayoría de los estudios se basan en procedimientos fenotípicos relativamente simples, como la medición de la glucemia en ayunas, lo que no revela adecuadamente la etiología subyacente.Por lo tanto, son muy importantes los esfuerzos para mejorar el fenotipo de la enfermedad.
El mejor método actual para identificar los genes que contribuyen a las enfermedades poligénicas son los estudios de asociación de todo el genoma, basados en la asociación de variantes genéticas comunes─polimorfismos de un solo nucleótido─con un fenotipo determinado, como la hiperglucemia. Hasta la fecha, en estudios de grandes cohortes se han encontrado más de 70 loci de genes que se asocian con la DM2, los que en su mayoría están implicados en la función de las células β.
Uno de los problemas en los estudios antes mencionados es el tamaño de las cohortes que deben ser estudiadas (a veces >100.000 personas) para generar suficiente poder estadístico. Tales cohortes son difíciles de fenotipificar con suficiente profundidad para revelar la compleja fisiología subyacente de la DM2. Así, inevitablemente, la mayoría de los estudios se basan en procedimientos fenotípicos relativamente simples, como la medición de la glucemia en ayunas, lo que no revela adecuadamente la etiología subyacente.Por lo tanto, son muy importantes los esfuerzos para mejorar el fenotipo de la enfermedad.
Un estudio reciente analizó la asociación de
37 loci de susceptibilidad de hiperglucemia con 3 rasgos clave que
influyen en la glucemia: sensibilidad a la insulina, procesamiento de la
insulina de las células β y, secreción de insulina. Esto reveló que los
loci de riesgo se agrupan en 3 grupos distintos, cada uno asociado
solamente con una de las 3 mediciones fenotípicas. Este estudio pone de
relieve tanto la marcada heterogeneidad fisiológica del rasgo glucémico
subyacente, y la necesidad de estratificar los fenotipos de la diabetes
para mejorar el poder de los estudios de asociación de todo el genoma.
Existe una necesidad urgente de nuevos enfoques para el estudio de las
interacciones entre el genotipo y el fenotipo en las células β.
Al respecto, ya se hicieron algunos avances en un nuevo estudio que analizó los islotes de donantes humanos con diferentes genotipos de riesgo. Este enfoque cuidadoso reveló la influencia directa de un subgrupo de loci de riesgo de diabetes sobre la secreción alterada de insulina ex vivo, y dio lugar a conceptos mecanicistas del papel de esas variantes genéticas.
¿Es posible un tratamiento personalizado de la diabetes tipio 2, basado en el genotipo?
En algunas enfermedades como el cáncer de mama, la genotipificación se hace en forma sistemática y se usa para predecir si el paciente se beneficiará de un fármaco específico. La genotipificación también ha revolucionado el tratamiento en algunos tipos de diabetes monogénica. El potencial para el tratamiento específico basado en el genotipo de la DM2 es menos claro, principalmente debido a que cada variante genética solo explica un pequeño grado de riesgo de la enfermedad.
Al respecto, ya se hicieron algunos avances en un nuevo estudio que analizó los islotes de donantes humanos con diferentes genotipos de riesgo. Este enfoque cuidadoso reveló la influencia directa de un subgrupo de loci de riesgo de diabetes sobre la secreción alterada de insulina ex vivo, y dio lugar a conceptos mecanicistas del papel de esas variantes genéticas.
¿Es posible un tratamiento personalizado de la diabetes tipio 2, basado en el genotipo?
En algunas enfermedades como el cáncer de mama, la genotipificación se hace en forma sistemática y se usa para predecir si el paciente se beneficiará de un fármaco específico. La genotipificación también ha revolucionado el tratamiento en algunos tipos de diabetes monogénica. El potencial para el tratamiento específico basado en el genotipo de la DM2 es menos claro, principalmente debido a que cada variante genética solo explica un pequeño grado de riesgo de la enfermedad.
Sin embargo, un estudio reciente reveló que
en los pacientes con DM2 portadores de una mutación del receptor
adrenérgico α2, el tratamiento con un fármaco contra este receptor
restauró la secreción de insulina. Esto aumenta la tentadora posibilidad
de menoscabar los estudios de asociación de todo el genoma para
fármacos nuevos dirigidos a desarrollar tratamientos personalizados para
grupos de individuos con subtipos específicos de DM2.
¿Existen otros tipos de células de los islotes involucradas en la patogénesis de la diabetes tipo 2?
Ha quedado bien establecido que la DM2 no está ocasionada simplemente por la falta de insulina y que la secreción inapropiada de glucagón por las células pancreáticas α representa un papel fundamental. El glucagón eleva la glucosa en sangre por estimulación de la gluconeogénesis y la salida de glucosa de los hepatocitos.
¿Existen otros tipos de células de los islotes involucradas en la patogénesis de la diabetes tipo 2?
Ha quedado bien establecido que la DM2 no está ocasionada simplemente por la falta de insulina y que la secreción inapropiada de glucagón por las células pancreáticas α representa un papel fundamental. El glucagón eleva la glucosa en sangre por estimulación de la gluconeogénesis y la salida de glucosa de los hepatocitos.
En la DM2 existe un marcado aumento de la secreción de glucagón
en presencia de glucemias elevadas, lo cual exacerba el efecto
hiperglucémico de la insulinopenia. También hay poca secreción de
glucagón en presencia de un tenor bajo de glucosa, lo cual puede
precipitar una hipoglucemia fatal.
Recientemente, el glucagón, un participante largamente ignorado en la homeostasis de la glucosa y la DM2, ha ganado su reconocimiento. El hallazgo espectacular de que la destrucción completa de las células ß por la estreptozotocina en los ratones con receptores de glucagón genéticamente anulados no provoca hiperglucemia y que el ratón de tipo salvaje contrae diabetes grave con la ablación de las células ß subraya la importancia del glucagón en la homeostasis de la glucosa.
Debido a que la expresión del receptor de glucagón en el hígado solo es suficiente para producir diabetes grave en el ratón sin receptor de glucagón con carencia de células ß funcionantes, la supresión de la producción de glucosa hepática inducida por el glucagón podría ser un buen objetivo terapéutico en la DM2.
Se ha propuesto a la metformina, muy utilizada para tratar la DM2 (especialmente en los obesos), para disminuir la glucemia por su antagonismo con la acción del glucagón, lo cual conduce a la inhibición de la producción de adenilciclasa y de AMPc, descendiendo de esta manera la gluconeogénesis hepática.
Recientemente, el glucagón, un participante largamente ignorado en la homeostasis de la glucosa y la DM2, ha ganado su reconocimiento. El hallazgo espectacular de que la destrucción completa de las células ß por la estreptozotocina en los ratones con receptores de glucagón genéticamente anulados no provoca hiperglucemia y que el ratón de tipo salvaje contrae diabetes grave con la ablación de las células ß subraya la importancia del glucagón en la homeostasis de la glucosa.
Debido a que la expresión del receptor de glucagón en el hígado solo es suficiente para producir diabetes grave en el ratón sin receptor de glucagón con carencia de células ß funcionantes, la supresión de la producción de glucosa hepática inducida por el glucagón podría ser un buen objetivo terapéutico en la DM2.
Se ha propuesto a la metformina, muy utilizada para tratar la DM2 (especialmente en los obesos), para disminuir la glucemia por su antagonismo con la acción del glucagón, lo cual conduce a la inhibición de la producción de adenilciclasa y de AMPc, descendiendo de esta manera la gluconeogénesis hepática.
Otras estrategias para reducir la acción del
glucagón incluyen la reducción de la liberación de glucagón de las
células pancreáticas α y el bloqueo de la estimulación del glucagón por
la producción de la glucosa hepática. De hecho, los antagonistas del
receptor del glucagón mejoran la glucemia en la DM2 como así se cree
que los miméticos del péptido símil glucagón y los inhibidores de de la
dipeptidil peptidasa 4 (DPP-4; la enzima que inactiva al péptido símil
glucagón 1) mejoran la homeostasis de la glucosa, al menos en parte,
mediante la reducción de los niveles del glucagón plasmático.
La reducción de la producción hepática de
glucosa también puede ser en parte la razón por la cual se puede
alcanzar el excelente control de la diabetes (antes de la pérdida
importante de peso) mediante una dieta muy baja en calorías.
Es urgente adquirir un mayor conocimiento de
los mecanismos reguladores de la secreción y acción del glucagón, tanto
en enfermedad como en la salud, y de cómo ese mayor conocimiento puede
servir para un mejor tratamiento de la DM2.
¿Se puede revertir la disfunción de las células β en la diabetes?
El UK Prospective Diabetes Study Group (UKPDS) demostró que existe una declinación inexorable de la función de las células β a través del tiempo, ya sea con el tratamiento con dieta, insulina o sulfonilureas. Una pregunta clave es cuál es la causa de esa declinación y si la misma puede ser revertida.
Se ha informado un mejoramiento de la secreción de insulina luego del tratamiento intensivo con insulina, mientras que una dieta hipocalórica puede mejorar la acción de la insulina, la función de las células β y la homeostasis de la glucosa en los pacientes con DM2. Por lo tanto, sería posible cierta reversión del deterioro de la función de las células β en esos pacientes, al menos en el corto plazo.
Los tratamientos farmacológicos actuales mejoran el control glucémico en la DM2 aumentando la secreción de insulina, lo que incluye las sulfonilureas, las cuales actúan cerrando los canales KATP.
¿Se puede revertir la disfunción de las células β en la diabetes?
El UK Prospective Diabetes Study Group (UKPDS) demostró que existe una declinación inexorable de la función de las células β a través del tiempo, ya sea con el tratamiento con dieta, insulina o sulfonilureas. Una pregunta clave es cuál es la causa de esa declinación y si la misma puede ser revertida.
Se ha informado un mejoramiento de la secreción de insulina luego del tratamiento intensivo con insulina, mientras que una dieta hipocalórica puede mejorar la acción de la insulina, la función de las células β y la homeostasis de la glucosa en los pacientes con DM2. Por lo tanto, sería posible cierta reversión del deterioro de la función de las células β en esos pacientes, al menos en el corto plazo.
Los tratamientos farmacológicos actuales mejoran el control glucémico en la DM2 aumentando la secreción de insulina, lo que incluye las sulfonilureas, las cuales actúan cerrando los canales KATP.
Los fármacos que mimetizan o amplifican la acción de las hormonas digestivas─conocidas como incretinas─también
mejoran la secreción de insulina. Por ejemplo, el péptido símil
glucagón 1es liberado en las células L intestinales, en respuesta a la
presencia de alimento en el intestino y, como otras incretinas, potencia
la secreción de insulina ante las concentraciones de glucosa que la
estimulan, pero no con contenidos bajos de glucosa. Esto hace del
péptido símil glucagón 1 una terapéutica dirigida atractiva porque
mejora la secreción de insulina solamente en respuesta a la comida,
cuando es necesaria, y no durante los intervalos entre las comidas,
cuando puede aumentar el riesgo de hipoglucemia.
Por lo tanto, en la actualidad, los
miméticos del péptido símil glucagón 1 y los inhibidores de DPP4 son
ampliamente utilizados y muy eficaces para estimular la secreción de
insulina y mantener la homeostasis de la glucosa en los pacientes con
DM2, sin que provoquen ni el aumento de peso ni el riesgo de hipoglucemia que acompañan al tratamiento con insulina y sulfonilureas.
Sin embargo, se han planteado interrogantes en cuanto a la seguridad
a largo plazo de las terapias basadas en la incretina: en particular,
por su efecto no deseado sobre las células ductales del páncreas
exocrino, lo que puede aumentar la incidencia de pancreatitis, que a su vez conduce potencialmente al cáncer de páncreas. Existe un debate acerca de si el riesgo potencial está justificado por los grandes beneficios de las incretinas.
Estudios recientes han revelado que la cirugía de bypass gástrico restaura rápidamente la homeostasis de la glucosa en los pacientes con DM2, antes de la pérdida importante de peso asociada al procedimiento. Una explicación para este hallazgo notable es que la cirugía provoca un aumento de la secreción del péptido símil glucagón 1 y de la liberación de insulina. Por lo tanto, la cirugía de bypass gástrico puede ser una terapia quirúrgica viable para el tratamiento de la DM2.
¿Dónde se puede esperar tener mayor conocimiento acerca de la falla de las células β en la diabetes tipo 2?
Como siempre, la investigación sobre la DM2 es un campo pendular y hay mucha energía dedicada a comprender más los cambios en la función de las células β que ocurren en la diabetes. Todavía sigue desconocida la causa básica del defecto en el acoplamiento estímulo-secreción en la DM2 y se requieren estudios más funcionales que utilicen islotes aislados de seres humanos con DM2.
Estudios recientes han revelado que la cirugía de bypass gástrico restaura rápidamente la homeostasis de la glucosa en los pacientes con DM2, antes de la pérdida importante de peso asociada al procedimiento. Una explicación para este hallazgo notable es que la cirugía provoca un aumento de la secreción del péptido símil glucagón 1 y de la liberación de insulina. Por lo tanto, la cirugía de bypass gástrico puede ser una terapia quirúrgica viable para el tratamiento de la DM2.
¿Dónde se puede esperar tener mayor conocimiento acerca de la falla de las células β en la diabetes tipo 2?
Como siempre, la investigación sobre la DM2 es un campo pendular y hay mucha energía dedicada a comprender más los cambios en la función de las células β que ocurren en la diabetes. Todavía sigue desconocida la causa básica del defecto en el acoplamiento estímulo-secreción en la DM2 y se requieren estudios más funcionales que utilicen islotes aislados de seres humanos con DM2.
En la actualidad, hay un renovado interés en
la desdiferenciación de las células β, investigada en modelos de ratón,
y en cómo se puede evitar o invertir, y en qué medida otras células de
los islotes pueden ser inducidas a diferenciarse en células β.
El papel del glucagón en la
DM2 también está recibiendo una considerable atención, desplegándose
aún más esfuerzos hacia una mejor estratificación de los fenotipos de la
enfermedad en los estudios genéticos.
Los avances terapéuticos recientes también apuntan a la posibilidad de restaurar la función endógena de las células β, aunque la estabilidad a largo plazo, la seguridad y la eficacia de estos enfoques todavía no se conoce. Se está a la espera que los resultados de todos estos estudios.
Traducción y resumen objetivo: Dra. Marta Papponetti
Los avances terapéuticos recientes también apuntan a la posibilidad de restaurar la función endógena de las células β, aunque la estabilidad a largo plazo, la seguridad y la eficacia de estos enfoques todavía no se conoce. Se está a la espera que los resultados de todos estos estudios.
Traducción y resumen objetivo: Dra. Marta Papponetti
No hay comentarios:
Publicar un comentario